

FDA Commissioner: "We Don't Validate AI Tools"
Once hailed as 'the most powerful government agency in the world,' the FDA has become an utterly irresponsible entity when it comes to AI safety, even allowing Babylon Health to operate unchecked.


🚨 Disclaimer: I conduct my own research and my own investigations using my own resources. Please ensure you cite my work responsibly.
Welcome to AI Health Uncut, a brutally honest newsletter on AI, innovation, and the state of the healthcare market. If you’d like to sign up to receive issues over email, you can do so here.
What was once the most powerful government agency in the world now looks sloppy and ineffective in the age of AI. Even worse, it refuses to take responsibility.
The U.S. Food and Drug Administration (FDA) is meant to safeguard public health and ensure responsibility, especially when new technologies like Artificial Intelligence (AI) enter healthcare. But that’s not happening. Instead, the FDA’s approach to AI safety for patients and clinicians is appallingly hands-off.
At the 2024 HLTH conference in Las Vegas, FDA Commissioner Robert Califf, MD, essentially admitted as much. He emphasized that AI safety is mostly the responsibility of health systems, stating they need to “conduct continuous local validation of artificial intelligence tools to ensure they are being used safely and effectively.”
This statement is shocking. But as I break down in this article, the agency’s dysfunction, data, and a long history of revolving doors between the FDA and private industry make its failure on AI safety unsurprising.
To me, this is about shifting blame and avoiding accountability. Plain and simple. And that’s not what American taxpayers are paying the FDA to do.
As a side note, it’s bittersweet for me to report these atrocities at the FDA, as my experience with the FDA staff has actually been quite positive. When I was going through the process of registering my company’s AI tool, there was a lot of information and bureaucracy, as with any government-related process. However, I found the FDA staff very helpful and extremely responsive, especially for a government agency, guiding me through the process.
I want to share my concerns, and those of others, about the FDA in the hope that we can acknowledge them and make improvements. If any of my readers are politically connected and know a member of Congress who can assist with this goal, I’d greatly appreciate the help. Unfortunately, here on Long Island, we have a track record of electing representatives who don’t give a sh*t: George Santos and now Tom Suozzi. 😊
Here’s the TL;DR:
1. Even When the FDA Tried to Validate AI Devices, Sloppiness Prevailed
2. The FDA Has Spent More Time on AI Explainability Than on Ensuring AI Safety and Effectiveness
3. ⚠️ Warning: Health LLMs and AI Scribes Are Unregulated — None Are Registered with the FDA!
4. The FDA Turned a Blind Eye to ‘The Madoff of Digital Health’ — Now the Key (Alleged) Fraudster is Back!
5. The Revolving Door Between the FDA and the Industries It Regulates
6. This FDA Chief Regulated Medical Devices. His Wife Represented Their Makers.
7. Things Are Different in Europe
8. Time to Audit the FDA!
9. My Take
1. Even When the FDA Tried to Validate AI Devices, Sloppiness Prevailed
I cite the most prominent studies highlighting the FDA’s sloppy and irresponsible approach to AI safety.
🔹 In a study published in Nature Medicine, researchers at Stanford University reviewed 130 medical AI devices that were approved by the FDA at the time. They found that 126 of the 130 devices were evaluated using only previously collected data. This means no one assessed how well the AI algorithms function on patients in real-world settings with active human clinician input. Moreover, less than 13% of the publicly available summaries of these devices’ performances reported sex, gender, or race/ethnicity.
Sources:
Wu E, Wu K, Daneshjou R, Ouyang D, Ho DE, Zou J. How medical AI devices are evaluated: limitations and recommendations from an analysis of FDA approvals. Nat Med. 2021 Apr;27(4):582–584. doi: 10.1038/s41591–021–01312-x. PMID: 33820998.
Hadhazy A. Debiasing artificial intelligence: Stanford researchers call for efforts to ensure that AI technologies do not exacerbate health care disparities. Stanford News. May 14, 2021. https://news.stanford.edu/2021/05/14/researchers-call-bias-free-artificial-intelligence/ [Accessed November 12, 2024]

🔹 A new Nature feature, “The testing of AI in medicine is a mess. Here’s how it should be done,” highlights critical issues related to the lack of transparency and rigorous studies by the FDA in approving AI-powered medical algorithms. One of the significant points raised is that AI systems in medicine are often approved with minimal oversight, which raises concerns in a field that directly impacts patient health and outcomes. The article specifically mentions that between 2020 and 2022, only 65 randomized controlled trials of AI interventions were published. This small number reveals an insufficient volume of robust scientific research validating these technologies. Furthermore, the feature emphasizes that “relatively few developers are publishing the results of such analyses,” indicating a troubling trend where companies prioritize speed to market over rigorous scientific validation.
🔹 A recent study, “Not all AI health tools with regulatory authorization are clinically validated,” published in Nature Medicine, has brought attention to a concerning oversight in the regulation of AI medical devices. Conducted by a collaborative team from institutions such as the University of North Carolina School of Medicine and Duke University, the study examined the FDA authorization process for over 521 AI-enabled medical devices between 2016 and 2022. Shockingly, the researchers found that 43% of these devices did not undergo clinical validation, meaning they were not tested on real patient data to determine their safety and effectiveness.

The absence of clinical validation poses significant risks. While these devices are intended to assist in crucial healthcare functions—such as diagnosing abnormalities in radiology, assessing pathology slides, and predicting disease progression—there remains a lack of empirical evidence confirming their reliability in actual medical settings. The authors of the study emphasize the need for rigorous, transparent validation processes. Although FDA authorization may signal regulatory approval, it does not necessarily equate to the device being clinically proven.
Three types of validation were highlighted in the study: retrospective validation, prospective validation, and randomized controlled trials. Retrospective studies use historical patient data, whereas prospective studies provide stronger evidence by testing the AI’s performance on current, real-time patient data. Randomized controlled trials, considered the gold standard, account for variables that may affect outcomes. Yet, only a small fraction of the devices analyzed were evaluated using this method.
This research has sparked calls for the FDA to tighten regulations and for manufacturers to publish detailed validation results. The goal is to increase transparency and ensure that these AI tools truly enhance patient care without compromising safety.
🔹 The recent scoping review of 692 FDA-approved AI/ML-enabled medical devices, published in NPJ Digital Medicine, sheds light on alarming gaps in reporting demographic and socioeconomic diversity metrics in AI-powered medical devices. Here are some eye-opening statistics and findings from the study:
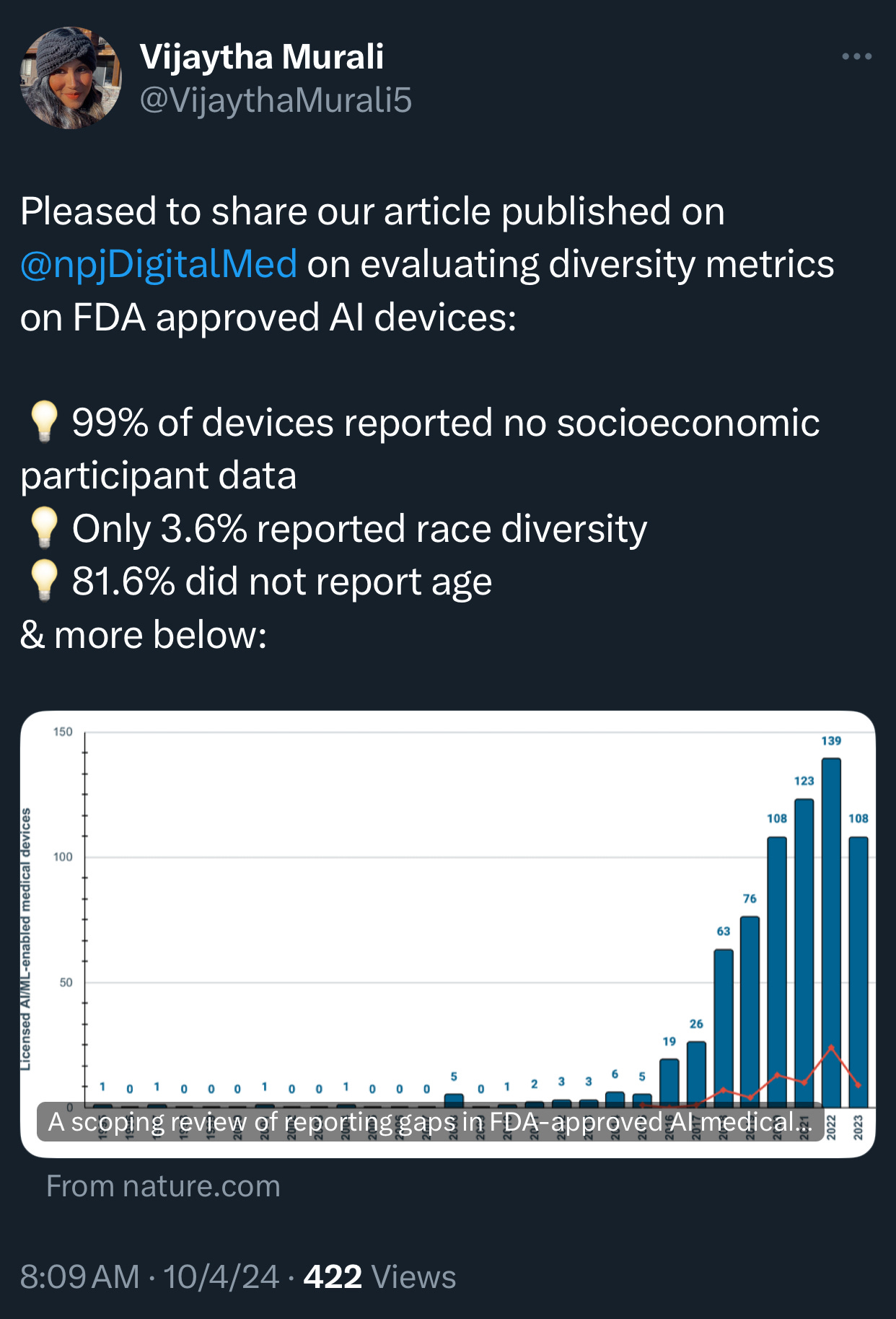
A staggering 99.1% of the AI medical devices failed to report any socioeconomic data on the participants involved in their testing. This omission leaves a vast gap in understanding the devices’ impact across different economic groups, which is crucial for assessing health equity implications.
Only 3.6% of the devices reported race or ethnicity information for the participants. This lack of data severely restricts the ability to evaluate whether these devices could exacerbate racial disparities in healthcare outcomes. The FDA’s oversight, in this case, appears insufficient to ensure that algorithmic bias doesn’t perpetuate existing inequities.
Age data reporting was absent in 81.6% of AI devices, meaning that age-related performance differences remain largely unexplored. Even though some devices have shown that algorithms trained on adult data often perform poorly on pediatric patients and vice versa, age inclusivity remains largely unaddressed.
Only 9.0% of AI devices had post-market surveillance data, further weakening the ability to assess their performance and impact in real-world clinical settings.
A mere 1.9% of the AI devices provided a link to a scientific publication detailing their safety and efficacy. This lack of transparency can hinder healthcare providers’ understanding of these tools’ reliability and potential risks.
Note: Post article’s publication, Dr. Hugh Harvey informed me that, despite the paper being peer-reviewed, several of its findings were erroneous. For a detailed review, please refer to Dr. Harvey’s full assessment here.
2. The FDA Has Spent More Time on AI Explainability Than on Ensuring AI Safety and Effectiveness
The FDA was expected to take the lead in regulating healthcare AI. Yet, navigating the labyrinthine complexities of AI in healthcare is no simple feat. The agency’s last significant action was on October 27, 2021, when it endorsed the 10 guiding principles of Good Machine Learning Practice (GMLP) in collaboration with Health Canada and the UK’s Medicines and Healthcare products Regulatory Agency (MHRA).
Critics argue the FDA has been overly preoccupied with AI explainability, sidelining patient-centric priorities like safety and effectiveness. The absence of rigorous clinical trials for AI-driven healthcare products has further fueled concern.
Some researchers believe that requiring AI explainability could stifle innovation. The insistence on understandable algorithms may degrade AI performance by forcing developers to use models that can be easily explained. Drs. Boris Babic and Sara Gerke highlight a flaw in the “post-hoc” explainability approach: when a “white box” model attempts to mirror the predictions of a “black box” AI using the output data rather than the original data, it offers little real insight into the AI’s inner workings. The authors suggest that “instead of focusing on explainability, the FDA and other regulators should closely scrutinize the aspects of AI / ML that affect patients, such as safety and effectiveness, and consider subjecting more health-related products based on artificial intelligence and machine learning to clinical trials.”
Sources:
Polevikov, S. Advancing AI in healthcare: A comprehensive review of best practices, Clinica Chimica Acta, Volume 548, Aug 2023, 117519, ISSN 0009-8981.
Babic B., Gerke S. Explaining medical AI is easier said than done. Stat News. July 21, 2021. https://www.statnews.com/2021/07/21/explainable-medical-ai-easier-said-than-done/ [Accessed November 12, 2024]
Babic B, Gerke S, Evgeniou T, Cohen IG. Beware explanations from AI in health care. Science. 2021 Jul 16;373(6552):284–286. doi: 10.1126/science.abg1834. PMID: 34437144.
3. ⚠️ Warning: Health LLMs and AI Scribes Are Unregulated — None Are Registered with the FDA!

None of the LLMs or AI scribes are registered with the FDA because they exploit the legal loophole provided by the 21st Century Cures Act. This act states that certain software and AI tools may be exempt from FDA regulatory approval if healthcare providers can independently review the recommendations and do not rely on them for diagnostic or treatment decisions. (Source: Encord.) That’s why so many of them exist — no scrutiny or randomized trials are necessary. Just a quick API connection to turbo-GPT-3.5 and integration with Google Voice, and boom, they’ve “invented” a “revolutionary” ambient medical AI transcribing system. (I discuss health AI liars and ‘AI tourists’ extensively in my articles, for example, here and here.)
4. The FDA Turned a Blind Eye to ‘The Madoff of Digital Health’ — Now the Key (Alleged) Fraudster is Back!
The SEC, the FBI, and the FDA all turned a blind eye to the alleged Babylon fraud, “The Madoff of Digital Health.” Despite clear FDA guidelines granting the agency power to exercise “enforcement discretion” over AI symptom checkers like Babylon’s, no action was taken. (Source: FDA Guidelines.)

From the get-go, Babylon’s CEO, Ali Parsa, opted not to register Babylon’s AI tool with the FDA, citing cost and time constraints. In reality, he feared rejection. Yet, the decision to bypass registration backfired, leading to greater costs. Every time a patient used the Babylon app, a “human in the loop” had to step in to legally ensure the patient didn’t follow the app’s advice blindly—one of the few measures in place to skirt FDA enforcement.
I’ve covered this extensively in my months-long investigation into Babylon’s alleged fraud, detailing 16 signs of potential fraud at Babylon Health and outlining 17 alleged lies by Ali Parsa.
Fun fact: As of yesterday, Ali Parsa has reemerged, pushing the same smoke-and-mirrors “AI algorithm” and asking for even more billions. His strategy seems to rely on investors having short memories, forgetting how he defrauded them for $1.5 billion, plus an extra sweet $1 billion wasted from U.S. taxpayer dollars through a capitated contract with the Centers for Medicare & Medicaid Services (CMS).
Remember: Theranos only pulled off a $700 million fraud, which looks like child’s play in comparison.
Unbelievable.

This is what happens when you don’t hold (alleged) fraudsters accountable the first time—they feel invincible and above the law.
Will Ali Parsa have the guts to set foot on American soil again? We shall see, but let’s hope the FBI, SEC, or FDA are paying attention. 🍿
5. The Revolving Door Between the FDA and the Industries It Regulates

Recent investigations by The British Medical Journal (BMJ) (here and here) have exposed disturbing financial ties between former FDA chiefs and the pharmaceutical industry. These findings cast serious doubt on the integrity and impartiality of the FDA’s decision-making process. The reports reveal that multiple FDA commissioners have transitioned into lucrative roles within the pharmaceutical sector after their tenure. This revolving door between the regulator and the industry it regulates generates glaring conflicts of interest, eroding public trust and damaging the FDA’s credibility.
6. This FDA Chief Regulated Medical Devices. His Wife Represented Their Makers.
As Jeffrey Shuren, MD, approaches his retirement after 15 years leading the FDA’s Center for Devices and Radiological Health (CDRH), a controversial New York Times investigation casts a shadow over his legacy. The report raises serious questions about potential conflicts of interest during his tenure. It centers on a specific issue: during Dr. Shuren’s time at the FDA, his wife, Allison, co-led a team of lawyers representing medical device manufacturers at Arnold & Porter, a prominent Washington law firm.

The New York Times reviewed thousands of court documents and FDA records, conducting numerous interviews and uncovering several instances where the Dr. Shuren’s roles appeared to intersect. The investigation reveals that while Dr. Shuren signed ethics agreements meant to prevent conflicts involving Arnold & Porter’s business, it remains unclear how rigorously these measures were enforced. An FDA spokesperson admitted to instances, dating back a decade, where Dr. Shuren should have recused himself or obtained ethics authorization to avoid any appearance of bias.
The allegations detailed in the New York Times article involve high-profile medical device companies, including Theranos, the discredited blood-testing startup, and Alcon, an eye-care company that was a client of Ms. Shuren and sought FDA approval for an implantable lens. The article also addresses other significant devices, such as breast implants. According to the report, Arnold & Porter “was working on a $63 billion acquisition of Allergan in 2019 when Dr. Shuren initially declined to push for a recall of Allergan’s breast implants, which were linked to a rare cancer.” Months later, Dr. Shuren and another FDA official reversed their stance and advocated for a recall, “citing additional reports of injuries and deaths related to the lymphoma.” While it’s not clear exactly what led Dr. Shuren to reverse his position, given his wife’s ties to Arnold & Porter, his about-face seems suspicious to say the least.
Sources:
Jewett, C. (2024, August 20). He Regulated Medical Devices. His Wife Represented Their Makers. The New York Times.
Jewett, C. (2024, September 25). Lawmakers Seek Inquiry of F.D.A. Device Chief’s Potential Conflicts. The New York Times.
7. Things Are Different in Europe
I hate to bring up other countries, as every nation has its own skeletons in the closet. However, European regulators don’t seem to be looking for scapegoats, at least not any longer. 😉 The EU Medical Device Regulation (MDR), in particular, holds AI manufacturers accountable for any issues their AI models produce. Enacted by the European Union in April 2017, the MDR enforces rigorous oversight on AI safety and validation standards. It classifies most healthcare AI as a “medical device,” requiring stringent testing, validation, and continuous monitoring before implementation. This regulation shifts much of the responsibility for AI accuracy and reliability to manufacturers. By holding AI providers to high standards, the MDR indirectly reduces risk for doctors relying on these technologies, though it does not eliminate liability entirely. Nevertheless, the MDR places substantial responsibility on AI developers to guarantee accuracy and safety, thereby lessening the burden on individual doctors to independently validate AI outputs. When errors occur, liability typically falls on manufacturers and developers due to the MDR’s strict regulations, offering doctors a buffer from direct accountability.
Moreover, the brand-new European Union AI Act (The EU AI Act of 2024) requires risk-based classifications of AI, particularly in healthcare, with stricter regulations for “high-risk” systems. This approach aligns closely with the conformity assessment procedures under the MDR.
8. Time to Audit the FDA!
Did you know that the FDA is one of the few government agencies subject to audits by independent oversight bodies like the Government Accountability Office (GAO)? This oversight is meant to drive accountability, enforce compliance with laws and regulations, and expose inefficiencies or risks of misconduct.
So, audit away! Conducting a thorough and transparent review of the FDA’s practices has never been more crucial.
9. My Take
The finger-pointing and responsibility-dodging are the real barriers to AI progress in healthcare. Healthcare already lags behind other industries by a decade or two in AI adoption. The potential of AI to save lives is enormous, but instead, we’re paralyzed by blame games and sidestepping.
Around the time ChatGPT launched, countless organizations, including the FDA, rushed to release “AI guidelines.” But let’s be honest: What good are guidelines if no one enforces them?
If the FDA doesn’t want to take the lead on AI safety, two critical questions arise.
First, I don’t care who leads—just take responsibility. If the FDA is unwilling or incapable of safeguarding AI use in healthcare, as indicated by various research findings, they should delegate that authority to someone else. We need a robust process and a dedicated entity to validate AI models’ safety in healthcare. AI is evolving at breakneck speed, but safety standards and best practices haven’t budged.
AI tools hold great promise for improving patient care. But until safety standards and best practices are in place, neither patients nor clinicians will be able to have full confidence in the tools’ quality, hampering adoption and impact. Worse, faulty tools may be put into practice, harming patients.
Second, if the FDA isn’t fulfilling its safety mandate—and there’s glaring conflict of interest involving a “revolving door” and corruption—why are American taxpayers footing a $7.2 billion annual bill to support the agency?
Over the years, I’ve published actionable recommendations like 12 Best Practices for AI in Healthcare and 6 Questions to Ask an AI Vendor aimed squarely at policymakers, regulators, and validation agencies. These weren’t written for clinicians or patients—I’m not expecting doctors to run GPU clusters to check for data contamination and overfitting. 😊
With over 20 years in machine learning and AI, I implore those in power: Use my expertise and recommendations.
Frankly, the FDA’s recent behavior does little to inspire confidence among healthcare professionals. Physicians are understandably skeptical of new technology, having endured disappointment and frustration from EHR systems for the past four decades.
The medical community needs confidence in regulatory bodies tasked with ensuring the safety of health technologies, especially AI. Yet, with the FDA, AHA, and AMA in bed with healthcare corporations, we all must do our own due diligence.
So maybe the FDA Commissioner is right: When it comes to AI safety, is it every person for themselves?
Is it time to reconsider the FDA’s leadership? Should the FDA be recalled?
Acknowledgment: This article owes its clarity, polish, and sound judgement to the invaluable contributions of Rachel Tornheim. Thank you, Rachel!
👉👉👉👉👉 Hi! My name is Sergei Polevikov. In my newsletter ‘AI Health Uncut’, I combine my knowledge of AI models with my unique skills in analyzing the financial health of digital health companies. Why “Uncut”? Because I never sugarcoat or filter the hard truth. I don’t play games, I don’t work for anyone, and therefore, with your support, I produce the most original, the most unbiased, the most unapologetic research in AI, innovation, and healthcare. Thank you for your support of my work. You’re part of a vibrant community of healthcare AI enthusiasts! Your engagement matters. 🙏🙏🙏🙏🙏
Thank you for your insights. I have followed Ali Parsa and Babylon Health since 2014 when he was pitching at Royal College of Medicine confabs. There were truck-sized holes in GP At Hand even then as it developed which then led to ditching the UK and opening big time in the US. His new venture Quadrivia is following the same track...and he started it less than a month after the entire Babylon Health business went bankrupt. (I'm still not sure what happened to the US assets.) I've written about it here with more of a focus on the financial backing and setup. https://telecareaware.com/babylon-healths-parsa-founds-new-ai-medical-assistant-venture-quadrivia-one-year-after-babylon-healths-failure/
The comments from the commissioner are interesting because this has NOT been what I've seen from the agency, with a number of companies going through the approvals process. Perhaps it depends on what team at the FDA you get, but it has been fairly rigorous, and although it's been... bumpy... some of the asks that they seem to now have are quite reasonable for balancing risk and patient benefit. For a while, I was seeing blanket denials for fairly flimsy reasons.
I do agree with the AI scribe thing being somewhat dubious, though the FDA has traditionally stayed out of a lot of these tools in the past, so I'm not really shocked.
I wouldn't be shocked, though so far that hasn't been my current experience of the agency—not to say that they don't have dumb things happening/getting approved, but I'm seeing fairly reasonable actors within it.